P07-27
Automated molecular modeling and property assessment for ADCs
Takashi IKEGAMI *
Life Science Dept., MOLSIS Inc.
( * E-mail: ikegami.takashi@molsis.co.jp )
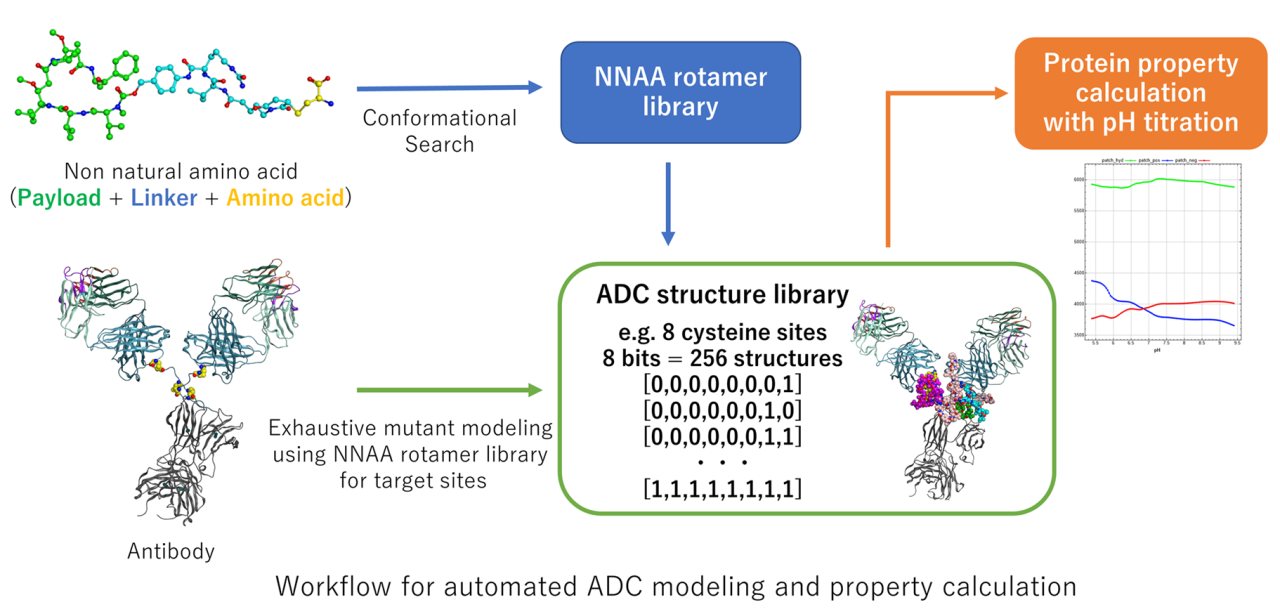
Antibody-drug conjugates (ADCs) are known as a class of key biopharmaceutical drugs in oncology. ADCs have a cytotoxic drug (payload) bonded to an antibody via a linker. Monoclonal antibody of ADC selectively binds to a target protein on the surface of a tumor cell and ADC delivers payloads into the tumor cell to cause cell death. In the development of ADCs, drugs are typically conjugated to solvent-exposed cysteine or lysine residues in the antibody. Because cysteine in antibodies is less common than lysine, cysteine is often used to minimize the distribution of the number and location of drugs. Since there are 8 cysteine residues in an antibody with inter-chain disulfide bonds, a total of 256 possible combinations of ADC structures can exist if chemical modifications are performed exhaustively. The composition ratio of the number of drug molecules conjugated to an antibody in an ADC is called the drug-antibody ratio (DAR). Drugs are often hydrophobic, and ADCs with high DAR can significantly alter their physical properties and cause experimental problems such as aggregation of ADCs. Therefore, it is important to predict ADCs with appropriate DAR among many combinations using in silico techniques. To estimate the effects in the physical properties of ADCs depending on the position and number of drugs, we propose a workflow that exhaustively model the 3D structure of ADCs and estimate their physical properties on Molecular Operating Environment (MOE)[1]. The workflow is shown in the figure:
1. Define amino acids covalently bound to the drug via the linker as non-natural amino acid (NNAA) and construct the side-chain rotamer libraries by conformational analysis.
2. Construct mutant library (ADC structure library) by replacing an amino acid (e.g. cysteine) with the NNAA at a specified position in the antibody. The stable side-chain conformation of NNAA is automatically selected from the rotamer library.
3. Calculate physical properties on the obtained ADC library, taking into account pH and conformational changes in ADCs.
This workflow will make it easier to find ADCs with the desirable physical properties and improve the efficiency of ADC design.
[1] Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2024.