P07-15
Acquisition of Bias Information for Protein-Ligand Docking by Mixed-Solvent Molecular Dynamics
Kaho AKAKI *, Keisuke YANAGISAWA, Yutaka AKIYAMA
School of computing, Institute of Science Tokyo
( * E-mail: akaki.k.aa@m.titech.ac.jp )
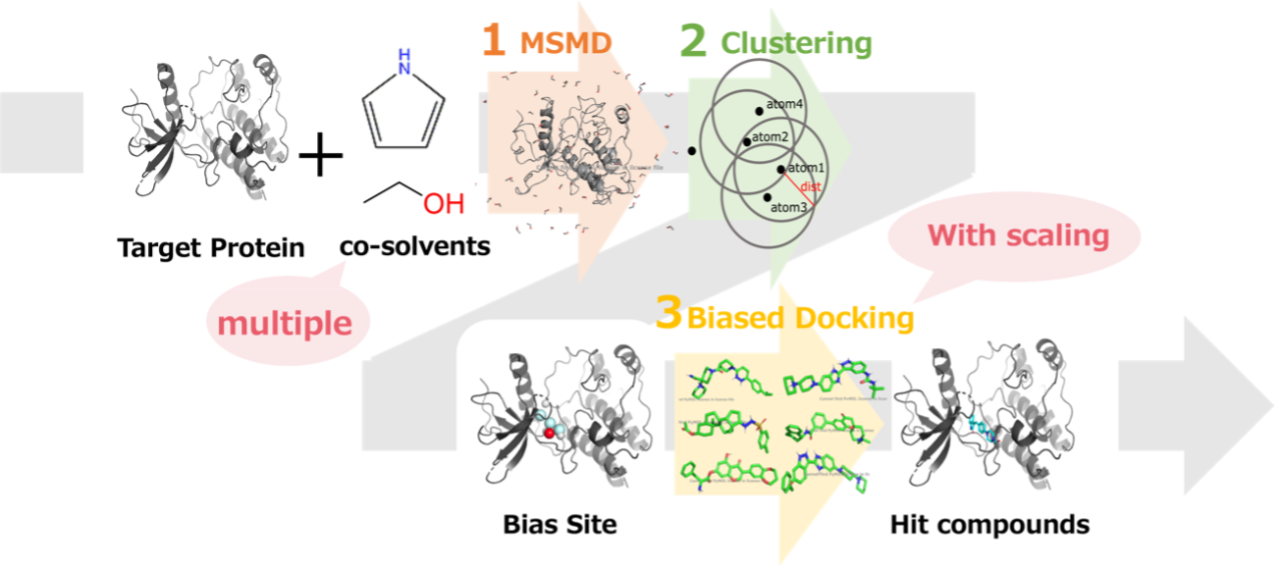
Virtual screening (VS) is a widely used method to computationally select drug candidates from a large number of compounds. In VS, protein-ligand docking is often performed to estimate binding affinities and binding modes. However, the accuracy of docking is not sufficient. Biased docking is a technique to introduce additional energy scores into docking aiming to improve the accuracy. One of the methods to estimate an adequate bias is mixed-solvent molecular dynamics, or MSMD. MSMD involve MD in the presence of explicit water molecules mixed with probe molecules or functional group fragments such as for hotspot detection, binding site identification, and binding free energy estimation. Arcon et al. shows that biased docking with interactions information from MSMD can provide better results [1]. However, they only used ethanol as a probe molecule.
In this study, we additionally utilized pyrrole as well as ethanol as probe molecules, and constructed four types of bias information: (1) aromatic rings, (2) hydrogen atoms to be hydrogen bond donors, (3) oxygen and nitrogen atoms to be hydrogen bond acceptors, and (4) nitrogen atoms that are not hydrogen bond acceptors. In addition, we introduced scaling for the strength of the bias. We evaluated our method on seven target proteins. Using MSMD, we calculated the interaction energies of multiple atom types and protein surfaces for every target, and obtained bias information for protein-ligand docking. The results indicated that the VS accuracy was improved for four of the seven target proteins with the bias obtained from ethanol and five of the seven target proteins with the bias obtained from pyrrole.
[1] Arcon JP, et al. JCIM, 59(8), 3572-3583, 2019.