P02-11
Binding Affinity Prediction Through Unsupervised Learning of Protein-Ligand MD Trajectories
Kodai IGARASHI *, Masahito OHUE
School of Computing, Institute of Science Tokyo
( * E-mail: igarashi@li.c.titech.ac.jp )
Prediction of binding affinity is an important technique that provides a useful basis for drug and enzyme design. However, prediction based on biochemical experiments and structural analysis requires much time and effort. Therefore, effective drug design is expected to be achieved by predicting binding affinity using bioinformatics.
Molecular dynamics simulation and machine learning have been proposed as common methods, but these methods have problems such as limited computation time and available data sets. There are also methods that use molecular dynamics simulations and deep learning to compare protein dynamics for a set of ligands for a specific protein and predict binding affinity. However, the computational cost of molecular dynamics simulations and deep neural networks is high.
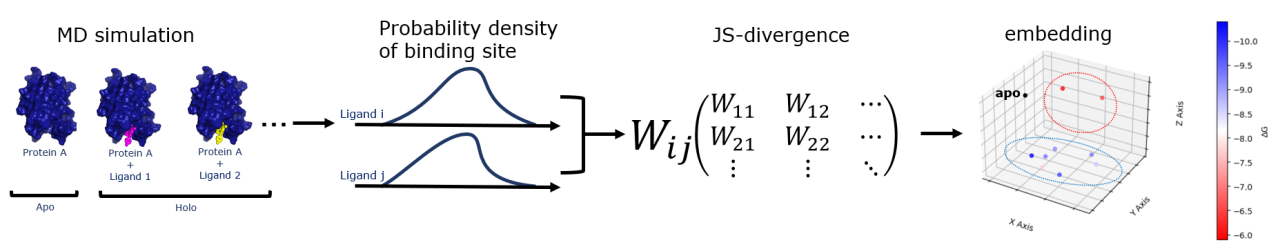
In this study, we propose a method to predict binding affinity for a specific protein by comparing protein dynamics using molecular dynamics simulations and Jensen-Shannon divergence, which reduces the computational cost of deep neural networks.
Specifically, we extract trajectory data of protein-ligand complexes generated by MD simulations and estimate the probability distribution of binding site residue trajectories. The similarity of the probability distributions among different ligands is then compared by Jensen-Shannon divergence. The points reflecting the obtained distance matrix are then projected into a two-dimensional space to represent the dynamics of the different ligands.
The results show that the accuracy of the proposed method is comparable to that of previous studies and reduces the computational cost by deep neural networks. We also investigated the effect of the initial structure used in the simulation on the simulation and on the prediction accuracy of binding affinity for the difference between the crystal structure and the modeling structure.