P02-08
Machine learning based prediction of quantum mechanical interaction energy between amino acid residues using fragment molecular orbital method
Tomohiro SATO *1, Watanabe CHIDURU1, Okiyama YOSHIO2
1Center for Biosystems Dynamics Research, RIKEN
2Graduate School of System Informatics, Kobe University
( * E-mail: tomohiro.sato@riken.jp )
Recently, various attempts for the application of quantum mechanics (QM) calculation to biomolecules were reported due to the development of methods to accelerate QM calculation for large molecular systems such as QM/MM or fragment molecular orbital (FMO) method [1, 2]. However, the computation cost is still high to apply QM calculation to high-throughput screening or molecular dynamics in which more than thousands of calculations were required. In this study, we created machine learning models to emulate interfragment interaction energies (IFIEs) calculation between amino acid residues by learning those data in FMO database (FMODB, URL: https://drugdesign.riken.jp/FMODB/) [3,4]. Thus , the model can be used as the alternative to conventional molecular force fields to evaluate inter/intra protein interactions for MD calculation or evaluation of antibody-antigen interaction.
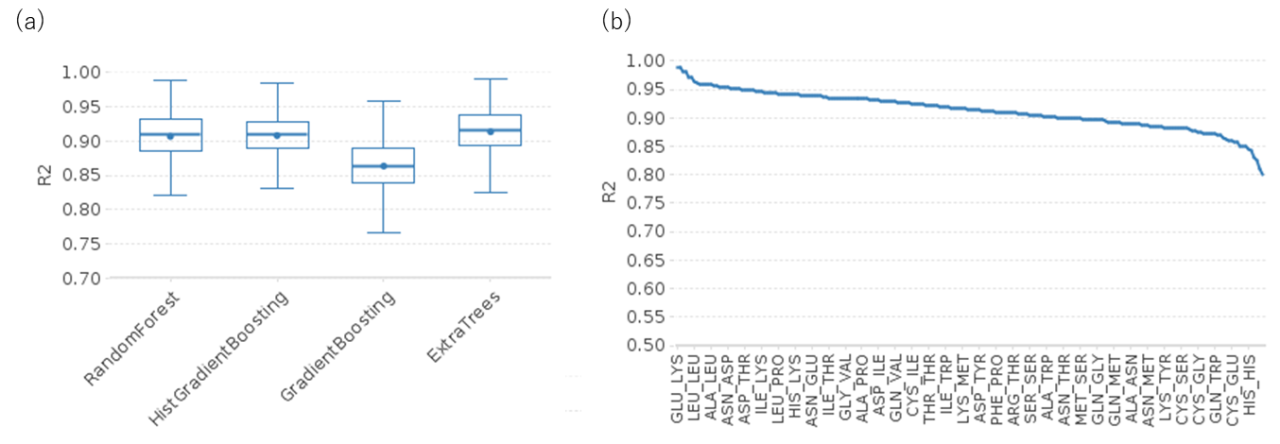
The regression models of IFIEs were built using random forest, extra trees, gradient boosting, and histogram-based gradient boosting based on the FMO dataset of 6,946 apoproteins, including 20,835 entries registered in FMODB. For each of the amino acid fragment pairs within 6 Å, which are not directly connected, the pairwise distances of heavy atoms in respective fragments were used as the explanatory variables to encode the geometric arrangement of the fragments, and learned with corresponding IFIEs . Among the machine learning techniques, the extra trees regressor recorded the highest prediction performance with R2 of 0.907 and RMSE of 2.032 in an average of all the 210 combinations of amino acid residues (Fig). The models provided excellent performances in amino acid pairs forming strong electrostatic interactions like glutamate-lysin pair and aspartate-lysin (R2=0.990, 0.989 by extra trees, respectively), and relatively low performances in cysteine-related pairs for which relatively small amount of structural data are available, such as non-disulfide bonded cysteine pair and cysteine-histidine pair (R2= 0.811, 0.800). Structural preparation procedure and consistency between the predicted IFIEs and their energy elements decomposed by PIEDA analysis are also to be assessed.
References
1. Warshel, A.; Levitt, M. J. Mol. Biol. 1976, 103, 227–249.
2. Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M., Chem. Phys. Lett. 1999, 313, 701–706.
3. Watanabe, C.; Watanabe, H.; Okiyama, Y.; Takaya, D.; Fukuzawa, K.; et al., CBIJ. 2019, 19, 5–18.
4. Takaya, D.; Watanabe, C.; Nagase, S.; Kamisaka, K.; Okiyama, Y.; et al., J. Chem. Inf. Model. 2021, 61, 777–794.