P01-17
High-precision and Efficient Prediction of Intermolecular Interaction Energies Using Deep Learning on Quantum Chemical Calculation Data
Yudai KOBAYASHI *, Natsuki KANAZAWA, Shigeyuki MATSUMOTO, Takao OTSUKA, Yasushi OKUNO
Department of Biomedical Data Intelligence, Graduate School of Medicine, Kyoto University
( * E-mail: kobayashi.yudai.72r@st.kyoto-u.ac.jp )
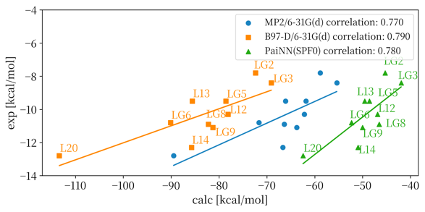
Understanding local interactions between drug molecules and target proteins is crucial in life sciences. While quantum mechanics (QM) calculations offer precise insights, they are computationally expensive for large-scale systems or extensive compound screening. Recent advances in artificial intelligence, particularly deep learning applied to QM calculations, have shown promise in addressing this challenge. We developed a QM-AI model based on PaiNN [1], an improved version of SchNet [2], trained on a neutral subset of the Solvated Protein Fragments (SPF) dataset [3], to predict intermolecular interaction energies in protein-ligand systems, specifically applying it to FK506-binding protein and ten ligand compounds. Figure 1 shows the correlations between the experimental binding affinities and the computed interaction energies. Our PaiNN(SPF0) model (green triangles) showed comparable performance to previous QM calculations using multilayer fragment molecular orbital (FMO) method by FMO2-MP2/6-31G(d) (blue circles) and FMO2-B97-D/6-31G(d) (orange squares) [4]. The correlation coefficients between the experimental and the computed values were R=0.780 for PaiNN(SPF0), R=0.790 (MP2), and R=0.770 (B97-D) for the QM calculations. These results demonstrate that our PaiNN(SPF0) model can predict FKBP-ligand interactions with accuracy comparable to QM calculations. It's remarkable that our model, trained on small multi-molecular systems, can effectively represent complex many-body interactions in large protein-ligand systems. Next, we focused on the computational efficiency of our QM-AI model. Typically, the QM computational times increase exponentially with the system size. Previous studies have indicated that the QM computational times for FKBP-ligand interactions were approximately 6 hours for the smallest ligand, LG2 (1712 atoms), and about 30 hours for the largest ligand, L20 (1792 atoms). In contrast, our QM-AI model, PaiNN(SPF0), has computed the FKBP-ligand interaction energies in 5.33 seconds for FKBP-LG2 and 5.76 seconds for FKBP-L20, demonstrating remarkable computational efficiency for such protein-ligand systems. Our proposed QM-AI model demonstrates the potential for rapid and efficient calculation of intermolecular interaction energies in protein-ligand systems.
References:
1. Schütt, K. T., Unke, O. T. & Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra. (2021).
2. Schütt, K. T. et al. SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. (2017).
3. Unke, O. T. & Meuwly, M. PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. Journal of Chemical Theory and Computation 15, 3678–3693 (2019).
4. Otsuka, T., Okioto, N., Taiji, M., Assessment and Acceleration of Binding Energy Calculations for Protein-Ligaind Complexes by the Fragment Molecular Orbital Method. J. Comput. Chem., 36, 2209-2218 (2015).