P01-03
Investigation of the utility of steered MD in the prediction of binding affinity: a case study of HSP90
Chisato KANAI *1, Enzo KAWASAKI1, Atsushi YOSHIMORI2
1INTAGE Healthcare Inc.
2Institute for Theoretical Medicine, Inc.
( * E-mail: kanai@intage.com )
In recent years, various models for de novo design using AI-generated compound structures have been proposed and widely used in drug discovery research. We offer Deep Quartet, a service that designs compound structures using a pharmacophore model based on the three-dimensional structure of proteins as the scoring function. Deep Quartet has been applied to various proteins where a large number of compound structures has been generated [1].
Even after applying ADME filters such as the Rule of 5 and synthetic accessibility scoring, thousands of compounds often remain as candidates. This situation is believed to be common to other de novo design systems using generative AI.
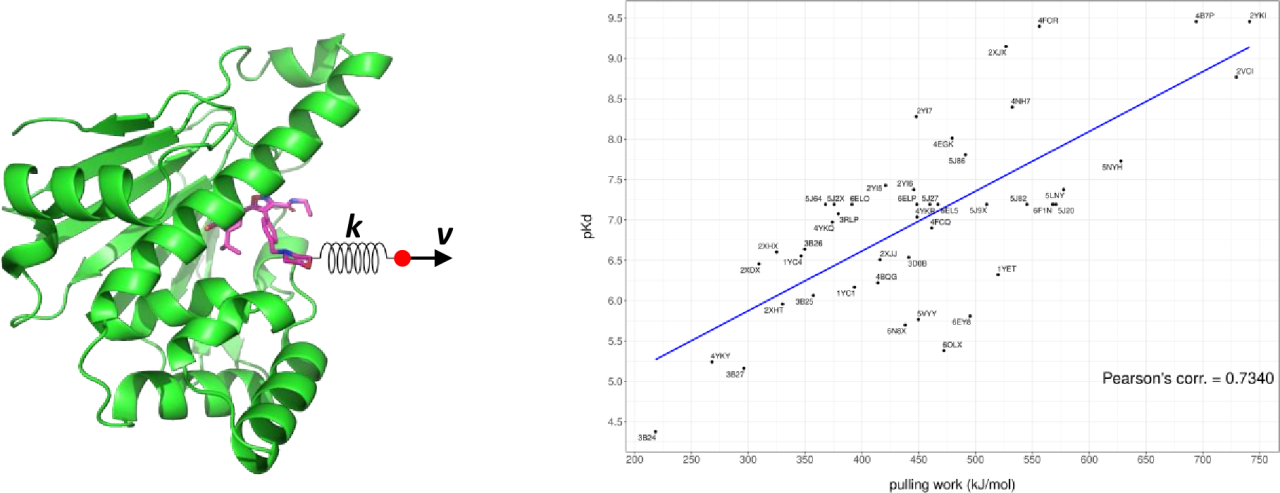
In actual drug discovery research, the number of in silico-designed compounds that can be synthesized is, typically, only a few, at most a few dozen. With this in mind, we investigated whether Steered MD could be used as a final selection method from the thousands of compounds designed by generative AI. Steered MD simulates the process of pulling a ligand out of its bound state with a protein using MD simulations, calculating the work energy required to dissociate the ligand from the protein. Although the results are from non-equilibrium MD simulations, previous studies [2, 3] have reported some correlation between ligand pulling work and binding free energy. An advantage of Steered MD is its significantly lower computational cost compared to binding free energy methods (FEP, TI, Umbrella Sampling, MMPBSA). Even if the prediction accuracy of the binding strength is slightly reduced, the low computational cost is still attractive, making it feasible to perform calculations for over a thousand compounds. In this poster presentation, we report the results of our evaluation of the binding strength of compounds based on Steered MD, using the protein HSP90 as an example.
[1] YOSHIMORI, Atsushi, et al. Design and synthesis of DDR1 inhibitors with a desired pharmacophore using deep generative models. ChemMedChem, 2021, 16.6: 955-958.
[2] VUONG, Quan Van; NGUYEN, Tin Trung; LI, Mai Suan. A new method for navigating optimal direction for pulling ligand from binding pocket: application to ranking binding affinity by steered molecular dynamics. Journal of chemical information and modeling, 2015, 55.12: 2731-2738.
[3] HO, Kiet; TRUONG, Duc Toan; LI, Mai Suan. How good is jarzynski’s equality for computer-aided drug design?. The Journal of Physical Chemistry B, 2020, 124.26: 5338-5349.