O08_01
Quantum Chemistry-Based Protein-Protein Docking without Any Empirical Parameters
Takeshi ISHIKAWA*
Graduate School of Science and Engineering, Kagoshima University
( * E-mail: ishi@cb.kagoshima-u.ac.jp)
Protein-protein interactions (PPIs) have recently garnered attention as prime targets for pharmaceuticals, making them one of the most vital research areas in the life sciences. Specifically, within the realm of protein science, significant efforts have been devoted to developing protein-protein docking methods to predict the three-dimensional structures of protein complexes. A vital aspect of protein-protein docking is the criterion for judging the appropriateness of the complex structure, which is also referred to as the scoring function, and most existing scoring functions utilize empirical parameters, such as molecular force fields.
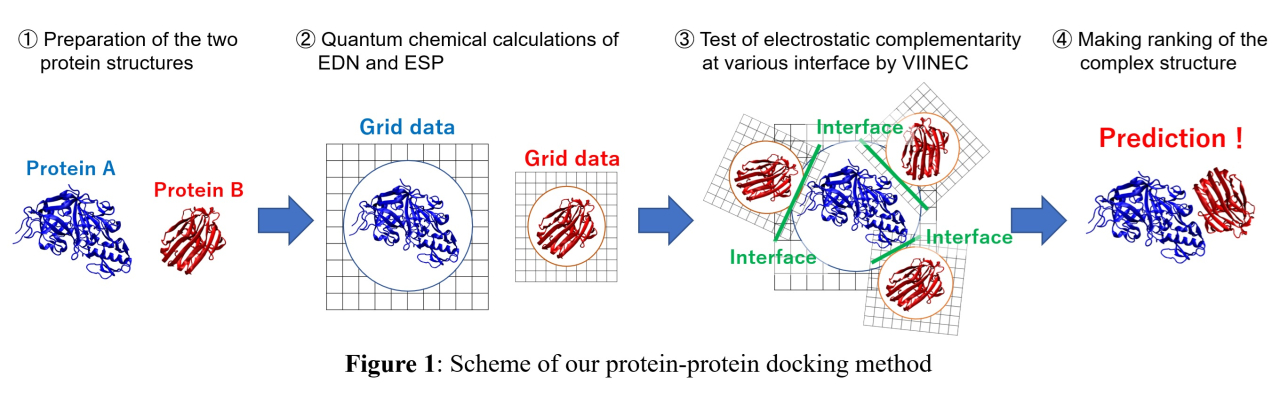
Recently, we developed "visualization of the interfacial electrostatic complementarity (VIINEC), " a method for analyzing PPIs based on full quantum chemical calculations[1-3]. In VIINEC, the interface of proteins within complexes is first identified using the electron density (EDN), following which electrostatic complementarity between the proteins is analyzed by plotting the electrostatic potential (ESP) over the interface. Applying VIINEC to 17 complexes found that ESP patterns of the two proteins at the interface are inverted in positive and negative and match each other like a puzzle. These results suggest that VIINEC can predict complex structures by seeking structures with high electrostatic complementarity.
This study developed a novel protein-protein docking approach based on VIINEC[4]. It can be regarded as a protein-protein docking without any empirical parameters as it solely relies on the EDN and ESP obtained from full quantum chemical calculations. The scheme of our protein-protein docking is given in Figure 1. Performance evaluations were conducted using 53 complexes in a benchmark set for protein-protein docking. The results demonstrated a success rate of 75.4% for dockings employing bound state structures and 17.0% for those employing unbound state structures. These performance metrics are comparable to those of existing docking methods.
[1] T. Ishikawa*, Chem. Phys. Lett., 761 (2020) 138103
[2] H. Ozono, T. Ishikawa*, J. Chem. Theory Comput., 17 (2021) 5600
[3] H. Ozono, K. Mimoto, T. Ishikawa*, J. Phys. Chem. B, 126 (2022) 8415
[4] S. Kousaka, T. Ishikawa*, J. Chem. Theory Comput., 20 (2024) 5164