O05_03
Computational exploration of bipolar disorder multi-omics data in the quest for novel drug targets
Flora R. AIGBE *, Kosuke HASHIMOTO, Kenji MIZUGUCHI
Laboratory for Computational Biology, Institute for Protein Research, Osaka University
( * E-mail: u383788b@ecs.osaka-u.ac.jp )
Bipolar disorder (BD) is a difficult to manage psychiatric mood disorder. Despite available management options, it is associated with higher mortality rates with increasing incidence. This is partly due to its obscure pathophysiology and consequent lack of adequate drug targets. Potential drugs for proposed new targets such as Ca2+-calmodulin-dependent protein kinase kinase-2 raise safety concerns due to cellular metabolic implications. The polygenic nature of BD is still under investigation; studies continue to implicate new genes. Progress has been slowed by the difficulty in modelling BD or obtaining relevant data. Computer-aided approaches have been limited by focus on single omics data type (per study), such as genomic or transcriptomic data alone. This study was designed to systematically evaluate publicly available multi-omics data to identify robust signatures that will aid in potential new target(s) identification
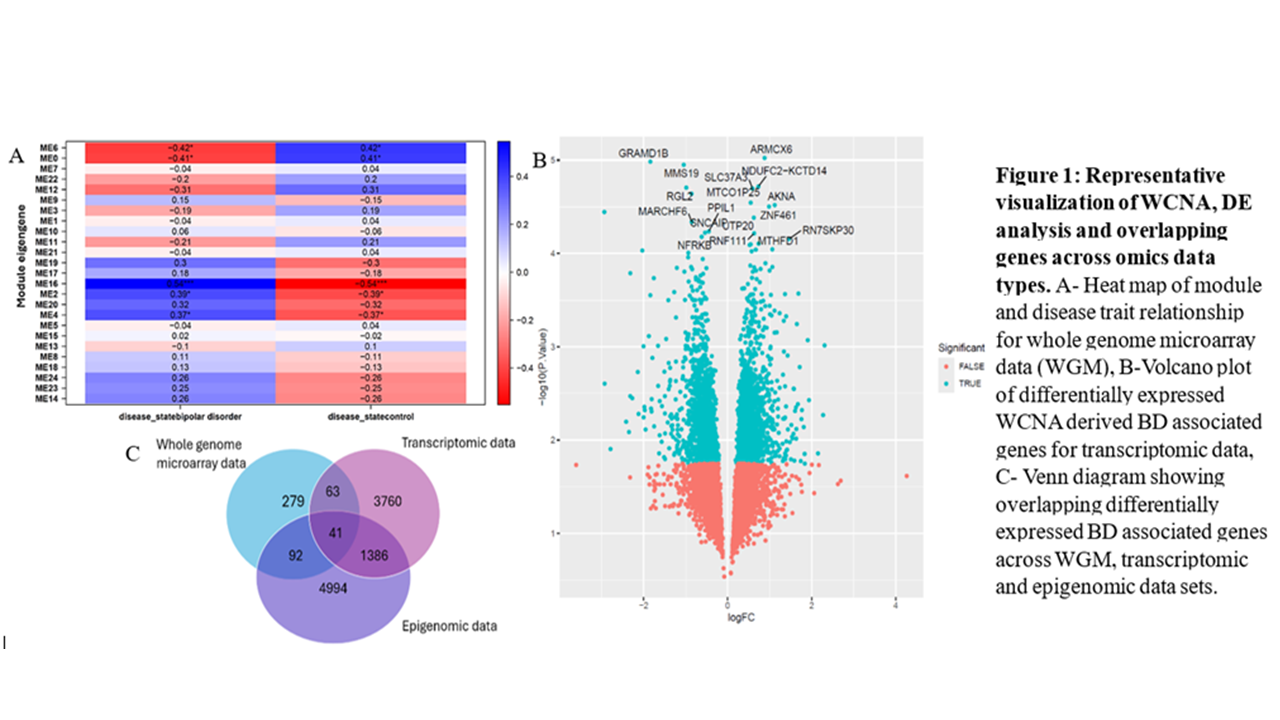
Whole genome microarray (GSE62191), RNAseq transcriptomic (GSE53239) and DNA methylation epigenomic (GSE112179) data were obtained from the Gene Expression Omnibus (GEO) database. These data sets were analyzed using R’s weighted gene co-expression network analysis (WGCNA) respectively. BD-associated genes or probes from this step were then used for differential expression analysis (DEA) using limma R package and subsequent evaluation to derive a list of DE genes that overlap across all the different data types, to be used for further analyses.
Using WGCNA, clusters or modules of significantly correlated (Pearson’s r ≥ 0.5; Student’s t-test p ‹ 0.05) co-expressed genes or probes with BD were identified from all the data sets. Of these genes, upregulated and downregulated DE genes were selected based on Benjamini-Hochberg’s adjusted P value less than 0.05 for each data type respectively. About 474 genes from the whole genome microarray data, 5,249 transcripts from the RNAseq transcriptome data and 6,512 gene-mapped CpG methylation sites, or their respective probes, were found to be differentially expressed in the BD group relative to the control group. These were then sorted for overlapping genes across all the data types. Of these, at least 1 unannotated probe and 40 genes were identified to overlap across all 3. Among the annotated 40 overlapping genes are ARHGEF10L, with a functionally related BD-associated gene, ARHGEF7, and DGKD, which has been previously linked to BD. Findings so far show the potential of WGCNA followed by DEA to help identify potential new players in BD across multiple independent data sets and aid new target identification.