O01_02
Computational Chemistry and Structure-Based Molecular Design for a Cyclic Peptide Drug Discovery Platform
Atsushi MATSUO*
Research Division, Chugai Pharmaceutical Co.,Ltd
( * E-mail: matsuoats@chugai-pharm.co.jp )
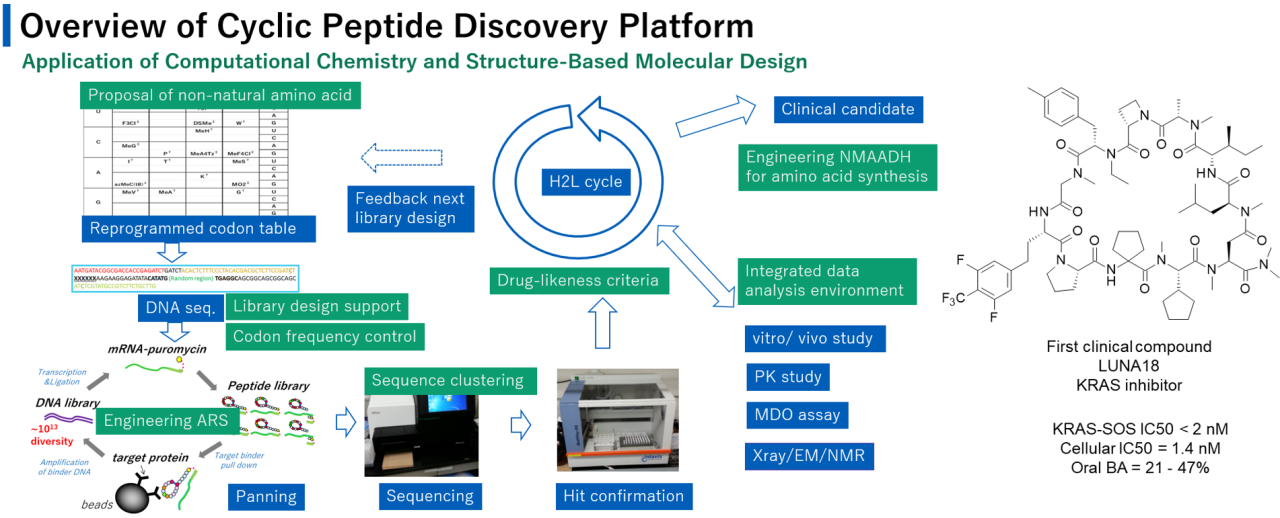
The construction of a technological platform for molecules inhibiting intracellular protein-protein interactions (PPIs) can be a game changer for drug discovery.
Although small molecules and antibodies are major modalities, they are not suitable for target proteins that lack a deep cavity for small molecule binding or proteins located in intracellular areas beyond the reach of antibodies. One potential solution is to use membrane-permeable mid-sized cyclic peptides (defined here as those with a molecular weight of 1000-2000 g/mol). We have established such a platform, and our most advanced molecule, the pan-RAS inhibitor LUNA181, is currently in clinical trials.
In this presentation, we will explain the details of the computational chemistry and structure-based molecular design used in the development of our cyclic peptide drug discovery platform2. We will also present new insights that have not yet been disclosed, such as detailed protein engineering, sequence clustering, and the development of a data analysis environment. Through this presentation, we will demonstrate that computational chemistry and structure-based molecular design are critical for the development of new drug modality platform technologies.
References
1) Tanada, M., et al., J. Am. Chem. Soc. 2023, 145, 16610
2) Ohta, A., et al., J. Am. Chem. Soc. 2023, 145, 24035